Biogeochemistry and FABM Coupling¶
pyGOTM integrates biogeochemical modelling through the Framework for Aquatic Biogeochemical Models (FABM) via its Python front-end, pyfabm. The coupling enables any FABM-compatible biogeochemical or ecological model to run within a pyGOTM single-column simulation without modifying the pyGOTM physics kernel.
What is FABM?¶
FABM (Framework for Aquatic Biogeochemical Models) is an open-source, model-agnostic coupling framework that allows biogeochemical models to be compiled once and coupled to different physical ocean and lake models without code changes. Each FABM model describes:
State variables — concentrations of biogeochemical quantities (e.g. phytoplankton, dissolved oxygen, nutrients).
Source/sink rates — the net production or consumption of each state variable due to biological reactions, chemistry, and air-sea exchange.
Dependencies — physical environment fields that the biogeochemical model needs from the host (temperature, salinity, light, density, etc.).
Diagnostics — derived quantities the model exposes back to the host (e.g. light attenuation, chlorophyll a).
pyfabm is the Python binding to the FABM shared library. It exposes the
FABM model described in a fabm.yaml configuration file as a
pyfabm.Model object whose state arrays, rates, diagnostics, and
dependencies can be read and written from Python. A wide range of models are
available — from simple nutrient-phytoplankton-zooplankton (NPZ) models to
complex biogeochemical models such as ERSEM and HBM-NEMO-PISCES.
Enabling FABM in pyGOTM¶
FABM is activated by adding a fabm block to the GOTM YAML configuration:
fabm:
use: true
config_file: fabm.yaml # path to the FABM model YAML, relative to gotm.yaml
freshwater_impact: true # dilute/concentrate tracers with precipitation
repair_state: false # clamp negative concentrations to zero
feedbacks:
shade: false # allow FABM to modify light attenuation
albedo: false # allow FABM to modify surface albedo
surface_drag: false # allow FABM to modify surface drag
The config_file key (also accepted as config, yaml, or file)
points to the FABM model YAML that specifies which biological models are
active. If not supplied, the default fabm.yaml in the same directory as
gotm.yaml is used.
The coupling is completely optional. If fabm.use: false (the default),
the physics-only compiled Numba loop runs unchanged and pyfabm is never
imported.
FABM output variables¶
When FABM is active, pyGOTM discovers output variables directly from the
instantiated pyfabm.Model. Every FABM state variable is written to the
NetCDF result, and every pyfabm diagnostic whose output flag is enabled is
written as well. Names use FABM’s output_name metadata, so slashes in model
paths are normalized to underscores. For example, gotm/npzd variables are
reported as npzd_phy and npzd_PAR, and bb/passive configured as
sed/c is reported as sed_c.
Interior state variables and interior diagnostics are written as
(time, z, lat, lon) profiles. Surface state, bottom state, and horizontal
diagnostics are written as (time, lat, lon) scalar fields. pyGOTM attaches
the FABM units and long_name metadata to each emitted NetCDF variable,
which makes the output schema and downstream integrations model-agnostic.
The host feedback placeholders surface_albedo,
surface_drag_coefficient_in_air, and
attenuation_coefficient_of_photosynthetic_radiative_flux remain present for
FABM-active runs for compatibility with existing validation reports. They are
separate from the model-discovered FABM namespace.
Use pygotm schema output --config path/to/gotm.yaml --json to inspect the
exact dynamic output surface before running a FABM-active configuration.
Installation Boundary¶
FABM coupling requires pyfabm from conda-forge. The project conda
environment includes it through pygotm-conda-env.yml. The Python package
does not expose a pygotm[fabm] pip extra because current FABM-capable
pyfabm builds are not available as matching PyPI dependencies. If FABM is
enabled without importable pyfabm, pyGOTM raises a clear runtime error
during FABM engine setup.
Coupling Architecture: Chunked Interleaved Loop¶
A fundamental constraint drives the design of the coupling:
Numba-compiled kernels cannot call back into Python, and pyfabm is a Python-level library. They cannot be interleaved at the sub-timestep level.
The solution adopted in pyGOTM is a chunk-based sequential coupling that interleaves physics and biogeochemistry at the granularity of chunks — contiguous windows of timesteps large enough to amortise the Python overhead while short enough to maintain tight coupling between the physical environment and the biological state.
Chunk size¶
The default chunk size is approximately one day of simulation time:
where \(\Delta t\) is the physics timestep in seconds. This is then rounded up to the nearest multiple of the output interval so that output boundaries are always aligned between physics and biogeochemistry.
The chunk size can be overridden by passing chunk_size to
integrate_gotm_compiled(). There is a natural
trade-off:
Smaller chunks — lower memory for the hydrodynamic state buffers, but more Python overhead.
Larger chunks — less Python overhead, but larger scratch arrays (shape \((N_\mathrm{chunk}+1) \times (N_\mathrm{lev}+1)\)). Memory grows as approximately \(7 N_\mathrm{chunk} N_\mathrm{lev}\) doubles (
float64arrays for T, S, ρ, h, nuh, radiation, and bottom stress).One-day default — balances memory and overhead across typical timesteps of 60–600 s.
The chunk size has no impact on numerical accuracy for the biological equations — the biogeochemistry is advanced at the same timestep \(\Delta t\) as the physics within each chunk; chunking only determines how often the stored hydrodynamic snapshots are refreshed.
Per-chunk execution sequence¶
Each chunk k covers timesteps \(k \cdot C \ldots k \cdot C + C - 1\) and executes two sequential passes.
flowchart TD
start(["Chunk k — timesteps k·C … k·C + C − 1"])
P["1 · Physics pass — Numba JIT\nrun_compiled_time_loop(nt=C, hydro_store=ON)"]
buf["Advances T, S, U, V, k, ε by C steps\nStores T, S, ρ, h, nuh, rad, τ_b at every step\n(hydro buffers · shape C+1 × nlev+1)"]
loop["2 · FABM pass — Python (for step = 0 … C)"]
fa["a. set_environment(T[step], S[step], ρ[step], …)"]
fb["b. sinking / rising advection (adv_center)"]
fc["c. turbulent diffusion (diff_center)"]
fd["d. biogeochemical rates (engine.get_rates)"]
fe["e. apply source/sink rates: cc += dt · rates"]
done(["cc — updated biological state passed to chunk k+1"])
start --> P --> buf
buf -->|"hydro snapshots"| loop
loop --> fa --> fb --> fc --> fd --> fe --> done
style start fill:#e8f4e8,stroke:#4a8f4a,color:#1a4a1a
style P fill:#dce8f7,stroke:#3a78b5,color:#1a4a7a
style buf fill:#dce8f7,stroke:#3a78b5,color:#1a4a7a
style loop fill:#fef3cd,stroke:#c8960c,color:#7a5c00
style fa fill:#fef3cd,stroke:#c8960c,color:#7a5c00
style fb fill:#fef3cd,stroke:#c8960c,color:#7a5c00
style fc fill:#fef3cd,stroke:#c8960c,color:#7a5c00
style fd fill:#fef3cd,stroke:#c8960c,color:#7a5c00
style fe fill:#fef3cd,stroke:#c8960c,color:#7a5c00
The biological state cc (shape n_vars × nlev) is passed from one chunk
to the next as a direct argument, providing continuity.
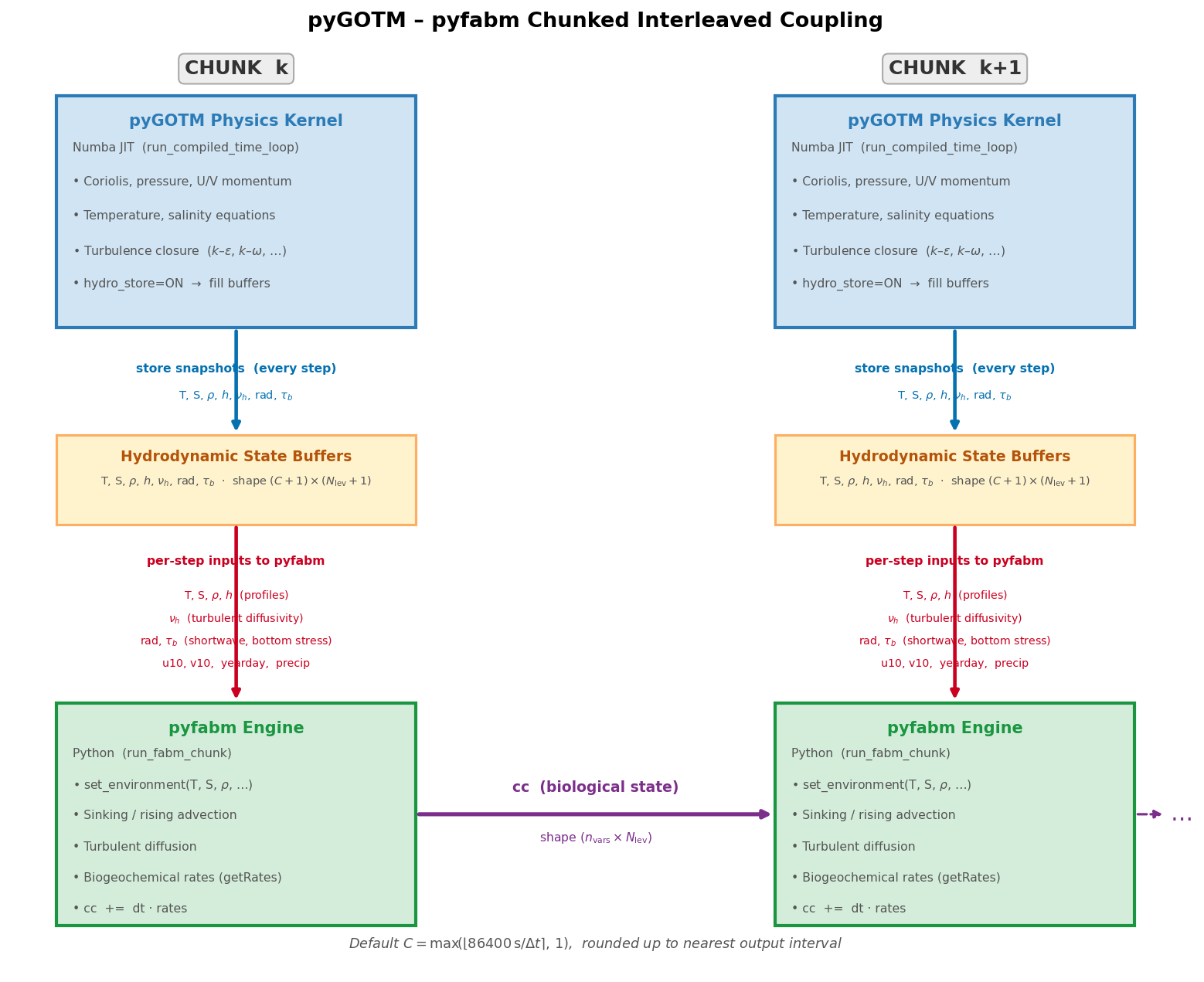
Figure — Chunked interleaved coupling between the pyGOTM Numba physics kernel and the pyfabm biogeochemical engine. Each chunk runs the full physics for \(N_\mathrm{chunk}\) timesteps, stores snapshots of the hydrodynamic state, then drives pyfabm through those same timesteps. Variables exchanged at each step are shown as labelled arrows.¶
Variables Exchanged¶
pyGOTM → pyfabm (dependencies)¶
At every biogeochemical timestep, pyGOTM sets the following FABM dependencies from the stored hydrodynamic snapshots:
FABM dependency name |
Shape |
Source in pyGOTM |
|---|---|---|
|
|
Potential temperature \(\theta\) [°C] — stored snapshot |
|
|
Practical salinity \(S\) [psu] — stored snapshot |
|
|
In-situ density \(\rho\) [kg m⁻³] — stored snapshot |
|
|
Layer thicknesses \(h_i\) [m] — stored snapshot |
|
|
PAR profile [W m⁻²] — computed from radiation snapshot, updated after light-feedback |
|
scalar |
Surface PAR [W m⁻²] — top-of-column value |
|
scalar |
Wind speed \(|\mathbf{u}_{10}|\) [m s⁻¹] from (u10, v10) forcing |
|
scalar |
Fractional day of year (yearday + secondsofday / 86400) |
|
scalar |
Bottom friction velocity \(\tau_b\) [Pa] — stored snapshot |
Dependencies that are not exposed by the particular FABM model are silently
skipped. A call to engine.unresolved_dependencies() reports any required
dependencies that have not been set after initialization.
pyfabm → pyGOTM (feedbacks and diagnostics)¶
FABM diagnostic / output |
Shape |
Effect in pyGOTM |
|---|---|---|
|
|
Bio-shading: modifies the PAR profile fed back to FABM at the next rates call within the same timestep (light-feedback loop) |
Source/sink rates (all state variables) |
|
Applied to biological concentrations: |
Surface exchange rates |
|
Added only to the top layer (index |
Bottom exchange rates |
|
Added only to the bottom layer (index |
Vertical movement velocities |
|
Sinking or rising advection before the diffusion/reaction step |
Within-Chunk FABM Timestep¶
Each biological timestep within a chunk applies the following operations in
order, which exactly mirrors the sequence used by gotm_fabm.F90 in the
reference Fortran GOTM implementation:
Set environment — copy the stored hydrodynamic snapshots into pyfabm dependencies (temperature, salinity, density, cell thickness, PAR, wind, year-day, bottom stress).
Vertical advection (sinking/rising) — interior state variables with non-zero vertical movement velocities are advected using the conservative P2-PDM scheme (
adv_center) with zero-flux boundary conditions at top and bottom. Face velocities are computed by thickness-weighted interpolation of cell-centre velocities to interfaces, replicating lines 1161–1199 ofgotm_fabm.F90.Turbulent diffusion — each interior state variable is diffused vertically using the implicit Crank–Nicolson solver (
diff_center) with the turbulent diffusivity \(\nu_h\) from the stored hydrodynamic snapshot. Precipitation dilution is applied as a Neumann surface boundary condition: \(F_\mathrm{surf} = -c_{n,\mathrm{sfc}} \cdot \text{precip}\).Biogeochemical rates — pyfabm
getRates()is called after the transport step so that reaction rates act on the post-transport concentrations. The rates are retrieved three times with different surface/bottom flags to separate bulk, surface, and bottom contributions:bulk_rates = engine.get_rates(surface=False, bottom=False) surf_rates = engine.get_rates(surface=True, bottom=False) bot_rates = engine.get_rates(surface=False, bottom=True)
Light feedback — if
attenuation_coefficient_of_photosynthetic_radiative_fluxis exposed by the model, the PAR profile is recomputed from the biological attenuation and fed back into FABM before a second rates call within the same timestep. This two-pass approach captures the immediate effect of phytoplankton self-shading on the light available for growth.Apply rates — concentrations are updated:
\[c_i^{n+1} = c_i^n + \Delta t \cdot R_i^\mathrm{bulk} + \Delta t \cdot \delta_{i,\mathrm{top}} \left(R_i^\mathrm{surf} - R_i^\mathrm{bulk}\right)\big|_\mathrm{top} + \Delta t \cdot \delta_{i,\mathrm{bot}} \left(R_i^\mathrm{bot} - R_i^\mathrm{bulk}\right)\big|_\mathrm{bot},\]where surface and bottom exchange contributions are concentrated in the top and bottom cells respectively. Surface and bottom state variables (e.g. sediment fractions exposed by some FABM models) are updated separately from their single-layer rate arrays.
PAR Profile and Light Feedback¶
The downwelling photosynthetically active radiation (PAR) at cell-centre depth \(z_k\) is computed from the surface radiation snapshot using a two-component exponential attenuation:
where \(I_0\) is the surface shortwave, \(A\) is the fraction absorbed
in the first attenuation component (light_A), \(g_2\) is the
background attenuation depth scale (light_g2), and
\(K_\mathrm{bio}(z_k) = \sum_{k'=k}^{N} k_{c,k'} \Delta h_{k'}\)
is the cumulative biological attenuation integrated downward from the surface
using the kc (or
attenuation_coefficient_of_photosynthetic_radiative_flux) diagnostic
exposed by the FABM model.
When light_g2 ≤ 0, light feedback is disabled and the PAR profile is
the stored GOTM radiation profile unchanged.
Memory Layout¶
The hydrodynamic state buffers allocated once per integration are
Array |
Shape |
Content |
|---|---|---|
|
|
Temperature snapshots (bottom→top, index 0 = initial condition) |
|
|
Salinity snapshots |
|
|
Density snapshots |
|
|
Cell-thickness snapshots |
|
|
Turbulent diffusivity snapshots |
|
|
Shortwave radiation snapshots (PAR source) |
|
|
Bottom stress snapshots (scalar per step) |
Row 0 of each buffer holds the state at the start of the chunk (or the
initial condition on the first chunk). Rows 1…C hold the state after each
physics timestep. The last column (index nlev) is the surface interface
value.
If the final chunk is smaller than the default chunk size, the buffers are reallocated to the smaller size so no stale data is used.
FABM NetCDF output buffers are allocated after pyfabm.Model has been
instantiated, because only the live model knows its state and diagnostic output
names. Memory for those buffers scales as
\(O(N_\mathrm{vars} N_\mathrm{out} N_\mathrm{lev})\) for interior variables
and \(O(N_\mathrm{vars} N_\mathrm{out})\) for horizontal variables. Physics-
only runs allocate no FABM output buffers.
See Also¶
FABM Coupling — full FABM module API reference (FABMConfig, FABMEngine, coupling, fabm_loop)
Physics Overview — global solution sequence and where biogeochemistry fits
Air–Sea Interaction — surface forcing that provides wind and radiation to the coupling